
This web page was created programmatically, to learn the article in its unique location you may go to the hyperlink bellow:
https://www.news-medical.net/news/20250724/Scientists-open-new-atlas-of-genetic-diversity-with-advanced-sequencing.aspx
and if you wish to take away this text from our web site please contact us
A landmark research harnesses long-read sequencing to disclose huge, beforehand undetected structural variations in human DNA, reshaping our understanding of genetics and illness potential.
Study: Structural variation in 1,019 diverse humans based on long-read sequencing
In a current research printed within the journal Nature, researchers investigated large-scale structural variants (SVs), complicated and poorly understood insertions, deletions, and rearrangements in DNA, utilizing next-generation ‘long-read’ sequencing. Their groundbreaking dataset comprised 1,019 people throughout 26 world populations. The research additional leveraged a novel graph-based analytical framework, permitting for the creation of over 107,000 sequence-resolved biallelic SVs, which the authors made open-access.
The high-resolution genomic investigation not solely considerably furthers our understanding of the true variety of human genetics but additionally progresses our identification and future administration of disease-causing genetic variants in sufferers.
Background
Biology textbooks usually depict the human genome as a linear string of three billion combos of A, T, G, and C – our DNA, the constructing blocks of our lives. The actuality, nevertheless, is much extra dynamic, with our DNA demonstrating large-scale structural variants (SVs)—deletions, duplications, insertions, and inversions of complete DNA segments.
Despite accounting for many base-pair (bp) variations between any two organisms and being main contributors to and modulators of human well being, they continue to be notoriously tough to review and poorly understood. Short-read sequencing, the predominant sequencing know-how of at this time, splices lengthy DNA segments into tiny fragments, that are then amplified. While efficient for small variants, these applied sciences wrestle to map complicated SVs, particularly massive insertions and multiallelic variable quantity tandem repeats (VNTRs), that are generally missed solely.
Consequently, a overwhelming majority of the human genome stays invisible to science and drugs, permitting probably curable genetic ailments to persist unabated. Long-read sequencing is a comparatively novel know-how that may learn for much longer, steady stretches of DNA, thereby overcoming short-read sequencing’s major SV-associated shortcoming. Harnessing this know-how may unlock this hidden portion of the human genome and the medical treasures that lie inside.
About the research
The current work does simply this: A consortium of researchers undertook an enormous, multinational mission to map SVs utilizing a globally various cohort. Study samples have been acquired from the 1000 Genomes Project (1kGP) and initially comprised 1,064 samples (lymphoblastoid cell traces).
Strict high quality management (QC) utilizing a mixture of DNA focus willpower (multimode microplate reader), DNA purity analysis (spectrophotometer), and DNA fragment size verification (Femto Pulse system) diminished the dataset to 1,019. This dataset comprised individuals from 26 distinct ancestries throughout Africa, the Americas, Europe, and East and South Asia.
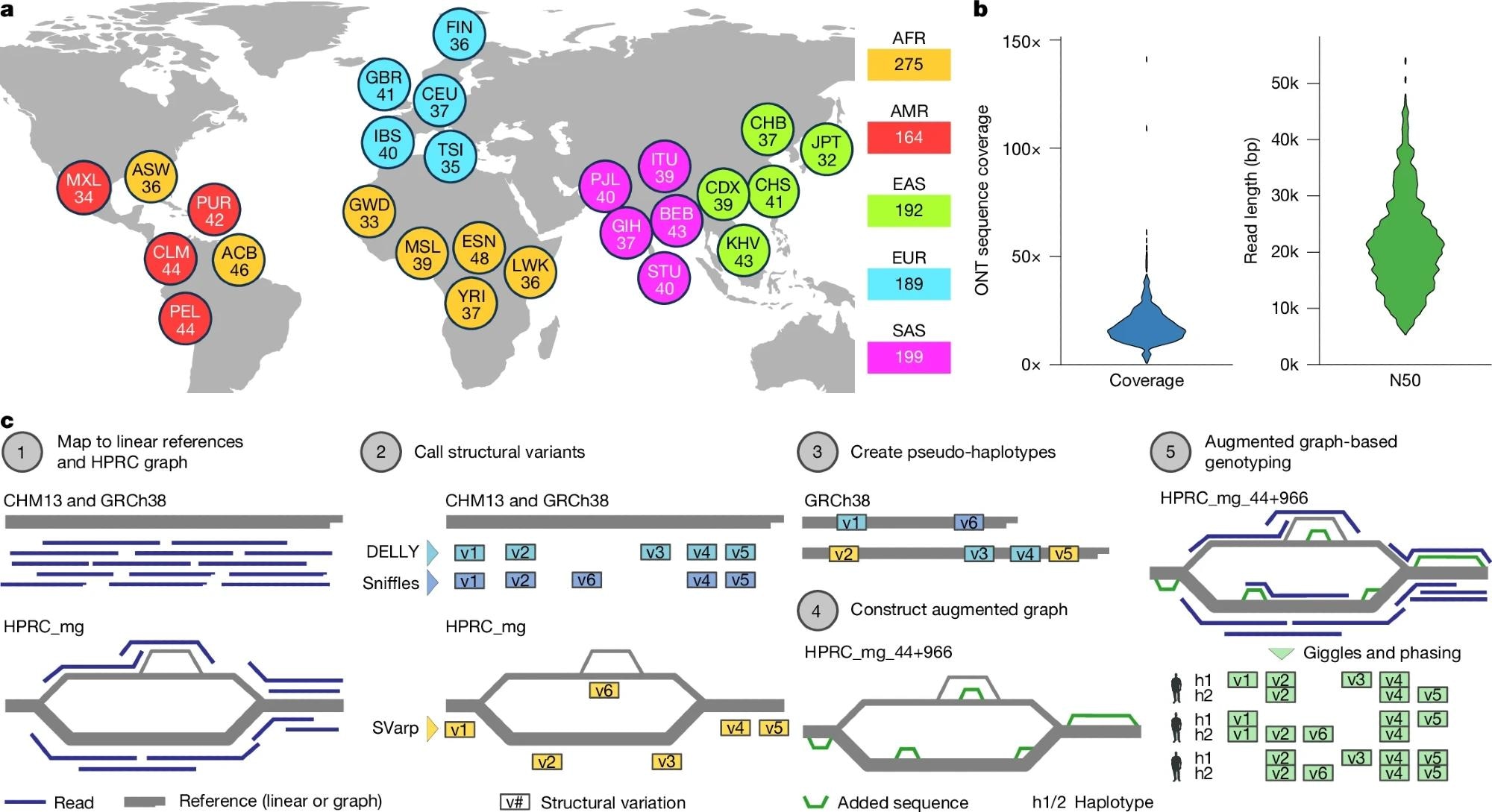 a, Breakdown of self-identified geographical ancestries for 1,019 long-read genomes representing 26 geographies (that’s, populations) from 5 continental areas. The three-letter codes used are equal to these used within the 1kGP section III18 and are resolved in Supplementary Table 2. b, ONT sequence protection per pattern, expressed as fold-coverage (left), and N50 learn size in base pairs (proper). c, Schematic of the SAGA framework for graph-aware discovery and genotyping of SVs utilizing a pangenome graph augmentation strategy. Basemap in a from Natural Earth knowledge (https://www.naturalearthdata.com).
a, Breakdown of self-identified geographical ancestries for 1,019 long-read genomes representing 26 geographies (that’s, populations) from 5 continental areas. The three-letter codes used are equal to these used within the 1kGP section III18 and are resolved in Supplementary Table 2. b, ONT sequence protection per pattern, expressed as fold-coverage (left), and N50 learn size in base pairs (proper). c, Schematic of the SAGA framework for graph-aware discovery and genotyping of SVs utilizing a pangenome graph augmentation strategy. Basemap in a from Natural Earth knowledge (https://www.naturalearthdata.com).
The long-read sequencing platform used was the Oxford Nanopore Technologies (ONT) LRS, a cutting-edge know-how able to producing knowledge with a median learn size of over 20,000 base pairs.
To analyze this complicated dataset, they engineered a novel computational framework known as SAGA (SV evaluation by graph augmentation). This course of concerned 4 key steps: First, aligning lengthy reads to each linear (GRCh38) and graph-based (HPRC) references; second, SV discovery utilizing Sniffles, DELLY, and the graph-aware SVarp algorithm, together with specialised remapping to resolve inversion alignment artifacts; third, augmenting the pangenome graph to include new SVs regardless of complexities in multiallelic VNTR genotyping; and eventually, genotyping the cohort utilizing Giggles software program to find out variant carriers (n = 967 samples), noting that multiallelic websites confirmed larger Mendelian inconsistency (15.1%).
Study findings
The current research resulted within the manufacturing of a richly annotated, publicly out there catalog of greater than 100,000 sequence-resolved SVs (biallelic), alongside 369,685 multiallelic variable quantity tandem repeats (VNTRs) genotyped utilizing the Vamos instrument. Identified SVs included inversions, deletions, duplications, and insertions, totalling a larger than tenfold improve within the variety of totally resolved insertion websites, filling a vital hole in human genomic data.
Mendelian consistency experiments leveraging household trios (two dad and mom and a baby) inside the cohort demonstrated the research’s excessive accuracy and very low error charge (deletions and insertions at simply 3.87% and 4.44%, respectively) for biallelic SVs. Notably, a lot of the novel SVs recognized on this research have been discovered to be extraordinarily uncommon, with 59.3% having a minor allele frequency (MAF) of lower than 1%. Individuals of African descent demonstrated the very best diploma of SV variety.
Finally, the research supplied novel insights into the organic mechanisms that create SVs, detailing how cell DNA components, equivalent to L1 and SVA retrotransposons, drive genetic innovation by selling SV formation and translocation by way of locus-specific processes, together with promoter hijacking (e.g., the 8q21.11 L1 supply aspect).
Conclusions
The current research represents a commendable leap ahead in our data and understanding of human genomics. The software of long-read sequencing efficiently allowed for the invention and annotation of extra SVs (particularly insertions), and the variety of the pattern cohort (26 distinct ancestries throughout a number of continents) validates the generalizability and world software of research findings.
Furthermore, the resultant complete and correct SV atlas, being open entry, opens the doorways to a brand new period of genetic drugs, permitting for the identification and early remedy of genetic situations that we hitherto did not even know existed. Notably, when utilized to rare-disease genomes, the useful resource filtered 55% of candidate SVs whereas retaining 94% (35/37) of validated causal variants. This open-access useful resource will probably be invaluable for the scientific group, enabling a deeper understanding of human evolution, inhabitants genetics, and the useful penalties of genetic variation.
Journal reference:
- Schloissnig, S., Pani, S., Ebler, J., Hain, C., Tsapalou, V., Söylev, A., Hüther, P., Ashraf, H., Prodanov, T., Asparuhova, M., Magalhães, H., Höps, W., Sotelo-Fonseca, J. E., Fitzgerald, T., Santana-Garcia, W., Moreira-Pinhal, R., Hunt, S., Pérez-Llanos, F. J., Wollenweber, T. E., … Korbel, J. O. (2025). Structural variation in 1,019 various people based mostly on long-read sequencing. Nature. DOI – 10.1038/s41586-025-09290-7,
This web page was created programmatically, to learn the article in its unique location you may go to the hyperlink bellow:
https://www.news-medical.net/news/20250724/Scientists-open-new-atlas-of-genetic-diversity-with-advanced-sequencing.aspx
and if you wish to take away this text from our web site please contact us


